Antioxidant supplements don't appear to have an impact on cerebrospinal fluid (CSF) biomarkers related to Alzheimer's disease, a clinical trial determined.
The combination of vitamin E, vitamin C, and alpha-lipoic acid did not lower levels of the amyloid and tau proteins that make up the plaques and tangles seen in the brain with Alzheimer's disease, Douglas R. Galasko, MD, of the University of California San Diego, and colleagues found.
The combination did reduce CSF levels of the oxidative stress biomarker F2-isoprostane by 19% but raised a safety concern with faster decline in cognitive scores, they reported online in the Archives of Neurology.
The popular antioxidant coenzyme Q (CoQ) had no significant impact on any CSF measures in the Alzheimer's Disease Cooperative Study antioxidant biomarker trial.
Oxidative damage is widespread in the brain in Alzheimer's disease and contributes to neuronal damage, Galasko's group explained.
Some prior observational evidence has pointed to lower Alzheimer's risk with an antioxidant-rich diet, although prevention trials with supplements have had mixed results, they noted.
Their study included 78 adults with mild to moderate Alzheimer's randomly assigned to double-blind treatment over 16 weeks with the combination of 800 IU vitamin E, 500 mg vitamin C, and 900 mg of alpha-lipoic acid once a day; CoQ alone at a dose of 400 mg three times a day; or placebo.
Vitamins C and E act as antioxidants by controlling dangerous free radicals produced when oxygen reacts with certain molecules, while alpha-lipoic acid spurs production of many antioxidant enzymes in the body. CoQ is an antioxidant that helps protect mitochondria from oxidation.
Serial CSF specimens collected from 66 of the participants showed only small changes from baseline.
Beta-amyloid 42, which accumulates to forms plaques in the Alzheimer's brain, declined by 8 pg/mL from a baseline of 190 pg/mL with the antioxidant combination and by 15 pg/mL from a baseline of 185 in the CoQ group, but neither was a significant difference from placebo.
Tau protein, which forms neurofibrillary tangles in the brain with Alzheimer's, fell by 23 pg/mL with the antioxidant combination from a baseline of 123 and by 9 pg/mL from a baseline of 109 in the CoQ group, but again neither differed from changes with placebo.
Levels of tau phosphorylated at a specific site (P-tau181) likewise declined slightly over the study period for the two antioxidant groups but without a significant difference from placebo.
The one significant change was in CSF levels of the oxidative marker F2-isoprostane, which is stable oxidized arachidonic acid.
The vitamin C and E plus alpha-lipoic acid group saw a 7 pg/mL reduction in F2-isoprostane from a baseline of 38 over the 16 weeks of treatment (P=0.04). The other groups showed no change.
"It is unclear whether the relatively small reduction in CSF F2-isoprostane level seen in this study may lead to clinical benefits in Alzheimer disease," the group cautioned.
Cognition, measured with the Mini-Mental State Examination, didn't improve in any of the groups. In fact, the decline in scores appeared accelerated in the antioxidant combination group, with a change of -4.6 points over the 16 weeks compared with -2.3 to -2.4 in the other two groups.
The researchers highlighted that as a potential safety concern that needs further careful assessment if longer-term trials are considered. The antioxidants were otherwise well tolerated.
Function, as measured on the Alzheimer's Disease Cooperative Study Activities of Daily Living Scale, didn't change in any group.
http://www.medpagetoday.com/Neurology/AlzheimersDisease/31721
Galasko D, et al "Antioxidants for Alzheimer disease: a randomized clinical trial with cerebrospinal fluid biomarker measures" Arch Neurol 2012; DOI:10.1001/archneurol.2012.85.
Tuesday 20 March 2012
Promising new research on Huntington's disease
Huntington's disease is a dreaded and debilitating congenital neurological disorder. There are little successful treatments and no cure. But a special type of brain cell forged from stem cells could help restore the muscle coordination deficits that cause the uncontrollable spasms characteristic of the disease.
"This is really something unexpected," Su-Chun Zhang, a University of Wisconsin-Madison neuroscientist said.
Zhang and his colleagues at the UW-Madison Waisman Center have learned how to make large amounts of GABA neurons from human embryonic stem cells, which they sought to test in a mouse model of Huntington's disease. The goal of the study, Zhang notes, was simply to see if the cells would safely integrate into the mouse brain. To their astonishment, the cells not only integrated but also project to the right target and effectively reestablished the broken communication network, restoring motor ability. It showed that locomotion could be restored in mice with a Huntington's-like condition.
What researchers found was intriguing, because GABA neurons reside in one part of the brain, the basal ganglia, which plays a key role in voluntary motor coordination. But the GABA neurons exert their influence at a distance on cells in the midbrain through the circuit fueled by the GABA neuron chemical neurotransmitter.
"This circuitry is essential for motor coordination," Zhang said, "and it is what is broken in Huntington patients. The GABA neurons exert their influence at a distance through this circuit. Their cell targets are far away."
That the transplanted cells could effectively reestablish the circuit was completely unexpected: "Many in the field feel that successful cell transplants would be impossible because it would require rebuilding the circuitry. But what we've shown is that the GABA neurons can remake the circuitry and produce the right neurotransmitter."
The study suggests that it may one day be possible to use cell therapy to treat Huntington's, but also because it suggests the adult brain may be more malleable than previously believed.
Zhang stresses that while the new research is promising; working up from the mouse model to human patients will take much time and effort. But for a disease that now has no effective treatment, the work could become the next best hope for those with Huntington's.
"This is really something unexpected," Su-Chun Zhang, a University of Wisconsin-Madison neuroscientist said.
Zhang and his colleagues at the UW-Madison Waisman Center have learned how to make large amounts of GABA neurons from human embryonic stem cells, which they sought to test in a mouse model of Huntington's disease. The goal of the study, Zhang notes, was simply to see if the cells would safely integrate into the mouse brain. To their astonishment, the cells not only integrated but also project to the right target and effectively reestablished the broken communication network, restoring motor ability. It showed that locomotion could be restored in mice with a Huntington's-like condition.
What researchers found was intriguing, because GABA neurons reside in one part of the brain, the basal ganglia, which plays a key role in voluntary motor coordination. But the GABA neurons exert their influence at a distance on cells in the midbrain through the circuit fueled by the GABA neuron chemical neurotransmitter.
"This circuitry is essential for motor coordination," Zhang said, "and it is what is broken in Huntington patients. The GABA neurons exert their influence at a distance through this circuit. Their cell targets are far away."
That the transplanted cells could effectively reestablish the circuit was completely unexpected: "Many in the field feel that successful cell transplants would be impossible because it would require rebuilding the circuitry. But what we've shown is that the GABA neurons can remake the circuitry and produce the right neurotransmitter."
The study suggests that it may one day be possible to use cell therapy to treat Huntington's, but also because it suggests the adult brain may be more malleable than previously believed.
Zhang stresses that while the new research is promising; working up from the mouse model to human patients will take much time and effort. But for a disease that now has no effective treatment, the work could become the next best hope for those with Huntington's.
Sunday 18 March 2012
Brain-derived neurotrophic factor (BDNF)
Brain-derived neurotrophic factor, also known as BDNF, is a secreted protein that, in humans, is encoded by the BDNF gene. BDNF is a member of the "neurotrophin" family of growth factors, which are related to the canonical "Nerve Growth Factor", NGF. Neurotrophic factors are found in the brain and the periphery.
BDNF acts on certain neurons of the central nervous system and the peripheral nervous system, helping to support the survival of existing neurons, and encourage the growth and differentiation of new neurons and synapses. In the brain, it is active in the hippocampus, cortex, and basal forebrain—areas vital to learning, memory, and higher thinking. BDNF itself is important for long-term memory. BDNF was the second neurotrophic factor to be characterized after nerve growth factor (NGF).
Although the vast majority of neurons in the mammalian brain are formed prenatally, parts of the adult brain retain the ability to grow new neurons from neural stem cells in a process known as neurogenesis. Neurotrophins are chemicals that help to stimulate and control neurogenesis, BDNF being one of the most active. Mice born without the ability to make BDNF suffer developmental defects in the brain and sensory nervous system, and usually die soon after birth, suggesting that BDNF plays an important role in normal neural development.
Click here for a list of research involving BDNF.
BDNF acts on certain neurons of the central nervous system and the peripheral nervous system, helping to support the survival of existing neurons, and encourage the growth and differentiation of new neurons and synapses. In the brain, it is active in the hippocampus, cortex, and basal forebrain—areas vital to learning, memory, and higher thinking. BDNF itself is important for long-term memory. BDNF was the second neurotrophic factor to be characterized after nerve growth factor (NGF).
Although the vast majority of neurons in the mammalian brain are formed prenatally, parts of the adult brain retain the ability to grow new neurons from neural stem cells in a process known as neurogenesis. Neurotrophins are chemicals that help to stimulate and control neurogenesis, BDNF being one of the most active. Mice born without the ability to make BDNF suffer developmental defects in the brain and sensory nervous system, and usually die soon after birth, suggesting that BDNF plays an important role in normal neural development.
Click here for a list of research involving BDNF.
Mice Study Suggests Caution with Alzheimer’s Drugs
As baby boomers become seniors, researchers are aggressively searching for medications that can reduce the detrimental effects of Alzheimer’s disease.
But a new study using mice suggests Alzheimer’s disease drugs now being tested in clinical trials may have potentially adverse side effects.
Northwestern University researchers discovered the drugs could act like a bad electrician, causing neurons to be miswired and interfering with their ability to send messages to the brain.
“Let’s proceed with caution,” said Robert Vassar, Ph.D., professor of cell and molecular biology at Northwestern University Feinberg School of Medicine. “We have to keep our eyes open for potential side effects of these drugs.”
Ironically, he said, the drugs could impair memory. The drugs in question are designed to inhibit BACE1, the enzyme Vassar originally discovered that promotes the development of the clumps of plaque that are a hallmark of Alzheimer’s. The BACE1 enzyme works by cutting up and releasing proteins that form the plaques. Thus, drug developers believed blocking the enzyme might slow the disease. However, in Vassar’s new study, he found BACE1 also has a critical role as the brain’s electrician. The enzyme maps out the location of axons, the wires that connect neurons to the brain and the rest of the nervous system, a process called axonal guidance.
Laboratory research involved studying genetically altered mice in which the BACE1 enzyme was removed. In doing this, Vassar discovered the animals’ olfactory system – used for the sense of smell — was incorrectly wired. The axons of the olfactory neurons were not wired properly to the olfactory bulb of the brain. The findings show the key role of BACE1 in axonal guidance. “It’s like a badly wired house,” Vassar said. “If the electrician doesn’t get the wiring pattern correct, your lights won’t turn on and the outlets won’t work.”
Studying the mechanism of the olfactory system is a good model for reviewing nerve or axonal wiring. If the axons aren’t being properly connected in the olfactory system, Vassar said, the problem likely exists elsewhere in the brain and nervous system. The hippocampus could be particularly vulnerable to BACE1 blockers, he noted, because its population of neurons is continually being reborn, which may play a role in forming new memories. The neurons need to grow new axons that in turn must connect them with new targets. Axonal guidance is a continuous need.
Despite the new findings, “It’s not all bad news,” Vassar noted. “These BACE1 blockers might be useful at a specific dose that will reduce the amyloid plaques but not high enough to interfere with the wiring. Understanding the normal function of BACE1 may help us avoid potential drug side effects.”
The findings, from the scientist whose original research led to the drug development, are published in the journal Molecular Neurodegeneration.
Northwestern University researchers discovered the drugs could act like a bad electrician, causing neurons to be miswired and interfering with their ability to send messages to the brain.
“Let’s proceed with caution,” said Robert Vassar, Ph.D., professor of cell and molecular biology at Northwestern University Feinberg School of Medicine. “We have to keep our eyes open for potential side effects of these drugs.”
Ironically, he said, the drugs could impair memory. The drugs in question are designed to inhibit BACE1, the enzyme Vassar originally discovered that promotes the development of the clumps of plaque that are a hallmark of Alzheimer’s. The BACE1 enzyme works by cutting up and releasing proteins that form the plaques. Thus, drug developers believed blocking the enzyme might slow the disease. However, in Vassar’s new study, he found BACE1 also has a critical role as the brain’s electrician. The enzyme maps out the location of axons, the wires that connect neurons to the brain and the rest of the nervous system, a process called axonal guidance.
Laboratory research involved studying genetically altered mice in which the BACE1 enzyme was removed. In doing this, Vassar discovered the animals’ olfactory system – used for the sense of smell — was incorrectly wired. The axons of the olfactory neurons were not wired properly to the olfactory bulb of the brain. The findings show the key role of BACE1 in axonal guidance. “It’s like a badly wired house,” Vassar said. “If the electrician doesn’t get the wiring pattern correct, your lights won’t turn on and the outlets won’t work.”
Studying the mechanism of the olfactory system is a good model for reviewing nerve or axonal wiring. If the axons aren’t being properly connected in the olfactory system, Vassar said, the problem likely exists elsewhere in the brain and nervous system. The hippocampus could be particularly vulnerable to BACE1 blockers, he noted, because its population of neurons is continually being reborn, which may play a role in forming new memories. The neurons need to grow new axons that in turn must connect them with new targets. Axonal guidance is a continuous need.
Despite the new findings, “It’s not all bad news,” Vassar noted. “These BACE1 blockers might be useful at a specific dose that will reduce the amyloid plaques but not high enough to interfere with the wiring. Understanding the normal function of BACE1 may help us avoid potential drug side effects.”
The findings, from the scientist whose original research led to the drug development, are published in the journal Molecular Neurodegeneration.
Researchers reveal how a single gene mutation interferes with the suppression of appetite, leading to obesity
The discovery offers clues about how to turn on brain sensitivity to leptin and insulin, hormones that turn off appetite
Washington, D.C. -- Researchers at Georgetown University Medical Center have revealed how a mutation in a single gene is responsible for the inability of neurons to effectively pass along appetite suppressing signals from the body to the right place in the brain. What results is obesity caused by a voracious appetite.
Their study, published March 18th on Nature Medicine's website, suggests there might be a way to stimulate expression of that gene to treat obesity caused by uncontrolled eating.
The research team specifically found that a mutation in the brain-derived neurotrophic factor (Bdnf) gene in mice does not allow brain neurons to effectively pass leptin and insulin chemical signals through the brain. In humans, these hormones, which are released in the body after a person eats, are designed to "tell" the body to stop eating. But if the signals fail to reach correct locations in the hypothalamus, the area in the brain that signals satiety, eating continues.
"This is the first time protein synthesis in dendrites, tree-like extensions of neurons, has been found to be critical for control of weight," says the study's senior investigator, Baoji Xu, Ph.D., an associate professor of pharmacology and physiology at Georgetown.
"This discovery may open up novel strategies to help the brain control body weight," he says.
Xu has long investigated the Bdnf gene. He has found that the gene produces a growth factor that controls communication between neurons.
For example, he has shown that during development, BDNF is important to the formation and maturation of synapses, the structures that permit neurons to send chemical signals between them. The Bdnf gene generates one short transcript and one long transcript. He discovered that when the long-form Bdnf transcript is absent, the growth factor BDNF is only synthesized in the cell body of a neuron but not in its dendrites. The neuron then produces too many immature synapses, resulting in deficits in learning and memory in mice.
Xu also found that the mice with the same Bdnf mutation grew to be severely obese.
Other researchers began to look at the Bdnf gene in humans, and large-scale genome-wide association studies showed Bdnf gene variants are, in fact, linked to obesity.
But, until this study, no one has been able to describe exactly how BDNF controls body weight.
Xu's data shows that both leptin and insulin stimulate synthesis of BDNF in neuronal dendrites in order to move their chemical message from one neuron to another through synapses. The intent is to keep the leptin and insulin chemical signals moving along the neuronal highway to the correct brain locations, where the hormones will turn on a program that suppresses appetite.
"If there is a problem with the Bdnf gene, neurons can't talk to each other, and the leptin and insulin signals are ineffective, and appetite is not modified," Xu says.
Now that scientists know that BDNF regulates the movement of leptin and insulin signals through brain neurons, the question is whether a faulty transmission line can be repaired.
One possible strategy would be to produce additional long-form Bdnf transcript using adeno-associated virus-based gene therapy, Xu says. But although this kind of gene therapy has proven to be safe, it is difficult to deliver across the brain blood barrier, he adds.
"The better approach might be to find a drug that can stimulate Bdnf expression in the hypothalamus," Xu says. "We have opened the door to both new avenues in basic research and clinical therapies, which is very exciting."
Washington, D.C. -- Researchers at Georgetown University Medical Center have revealed how a mutation in a single gene is responsible for the inability of neurons to effectively pass along appetite suppressing signals from the body to the right place in the brain. What results is obesity caused by a voracious appetite.
Their study, published March 18th on Nature Medicine's website, suggests there might be a way to stimulate expression of that gene to treat obesity caused by uncontrolled eating.
The research team specifically found that a mutation in the brain-derived neurotrophic factor (Bdnf) gene in mice does not allow brain neurons to effectively pass leptin and insulin chemical signals through the brain. In humans, these hormones, which are released in the body after a person eats, are designed to "tell" the body to stop eating. But if the signals fail to reach correct locations in the hypothalamus, the area in the brain that signals satiety, eating continues.
"This is the first time protein synthesis in dendrites, tree-like extensions of neurons, has been found to be critical for control of weight," says the study's senior investigator, Baoji Xu, Ph.D., an associate professor of pharmacology and physiology at Georgetown.
"This discovery may open up novel strategies to help the brain control body weight," he says.
Xu has long investigated the Bdnf gene. He has found that the gene produces a growth factor that controls communication between neurons.
For example, he has shown that during development, BDNF is important to the formation and maturation of synapses, the structures that permit neurons to send chemical signals between them. The Bdnf gene generates one short transcript and one long transcript. He discovered that when the long-form Bdnf transcript is absent, the growth factor BDNF is only synthesized in the cell body of a neuron but not in its dendrites. The neuron then produces too many immature synapses, resulting in deficits in learning and memory in mice.
Xu also found that the mice with the same Bdnf mutation grew to be severely obese.
Other researchers began to look at the Bdnf gene in humans, and large-scale genome-wide association studies showed Bdnf gene variants are, in fact, linked to obesity.
But, until this study, no one has been able to describe exactly how BDNF controls body weight.
Xu's data shows that both leptin and insulin stimulate synthesis of BDNF in neuronal dendrites in order to move their chemical message from one neuron to another through synapses. The intent is to keep the leptin and insulin chemical signals moving along the neuronal highway to the correct brain locations, where the hormones will turn on a program that suppresses appetite.
"If there is a problem with the Bdnf gene, neurons can't talk to each other, and the leptin and insulin signals are ineffective, and appetite is not modified," Xu says.
Now that scientists know that BDNF regulates the movement of leptin and insulin signals through brain neurons, the question is whether a faulty transmission line can be repaired.
One possible strategy would be to produce additional long-form Bdnf transcript using adeno-associated virus-based gene therapy, Xu says. But although this kind of gene therapy has proven to be safe, it is difficult to deliver across the brain blood barrier, he adds.
"The better approach might be to find a drug that can stimulate Bdnf expression in the hypothalamus," Xu says. "We have opened the door to both new avenues in basic research and clinical therapies, which is very exciting."
Sunday 11 March 2012
Deleting Nf1 Protein Quickens Relief from Depression
Eliminating a certain protein encourages the birth of new nerve cells and allows antidepressants to take effect more quickly, according to an animal study in The Journal of Neuroscience.
The normal role of the protein, called neurofibromin 1, is to prevent uncontrolled cell growth. The study suggests that treatment strategies developed to stimulate nerve cell birth may help treat depression more quickly, as current antidepressants typically take several weeks to take full effect.
Specifically, a particular section of the hippocampus produces new nerve cells during a process known as neurogenesis. This is made possible by specialized cells called neural progenitor cells (NPCs). Although previous research has shown that adult neurogenesis declines with age and stress, therapies known to alleviate symptoms of depression, such as exercise and antidepressants, increase neurogenesis.
A team of scientists, led by Luis Parada, PhD, of the University of Texas Southwestern, studied neurogenesis after removing the neurofibromin 1 (Nf1) gene from NPCs in adult mice. Results revealed that the removal of Nf1 increased the number and maturation of newborn nerve cells in the adult hippocampus.
Then, following seven days of antidepressant treatment, Nf1 mutant mice showed fewer depressive- and anxiety-like behaviors, whereas mice without the mutation took longer to show improvements.
“Our findings establish an important role for Nf1 in controlling neurogenesis in the hippocampus and demonstrate that activation of adult NPCs is enough to regulate depression and anxiety-like behaviors,” said study co-author Renee McKay, PhD, of the University of Texas Southwestern.
“Our work is among the first to demonstrate the feasibility of altering mood via direct manipulation of adult neurogenesis,” McKay added.
To determine if removing Nf1 leads to long-term changes in mice, the scientists ran 8-month-old mice through a variety of tests developed to measure anxiety- and depressive-like behaviors.
The mutant mice showed fewer anxious behaviors and also demonstrated resistance to the effects of chronic mild, unpredictable stress. Furthermore, even without antidepressants, removing Nf1 from NPCs in adult mice decreased symptoms of depression and anxiety.
“This study demonstrates that inducing neurogenesis is sufficient to produce antidepressant behavioral actions, and provides novel targets for therapeutic interventions,” said Ronald Duman, PhD, a neurogenesis expert from Yale University.
From PsychCentral.com
The normal role of the protein, called neurofibromin 1, is to prevent uncontrolled cell growth. The study suggests that treatment strategies developed to stimulate nerve cell birth may help treat depression more quickly, as current antidepressants typically take several weeks to take full effect.
Specifically, a particular section of the hippocampus produces new nerve cells during a process known as neurogenesis. This is made possible by specialized cells called neural progenitor cells (NPCs). Although previous research has shown that adult neurogenesis declines with age and stress, therapies known to alleviate symptoms of depression, such as exercise and antidepressants, increase neurogenesis.
A team of scientists, led by Luis Parada, PhD, of the University of Texas Southwestern, studied neurogenesis after removing the neurofibromin 1 (Nf1) gene from NPCs in adult mice. Results revealed that the removal of Nf1 increased the number and maturation of newborn nerve cells in the adult hippocampus.
Then, following seven days of antidepressant treatment, Nf1 mutant mice showed fewer depressive- and anxiety-like behaviors, whereas mice without the mutation took longer to show improvements.
“Our findings establish an important role for Nf1 in controlling neurogenesis in the hippocampus and demonstrate that activation of adult NPCs is enough to regulate depression and anxiety-like behaviors,” said study co-author Renee McKay, PhD, of the University of Texas Southwestern.
“Our work is among the first to demonstrate the feasibility of altering mood via direct manipulation of adult neurogenesis,” McKay added.
To determine if removing Nf1 leads to long-term changes in mice, the scientists ran 8-month-old mice through a variety of tests developed to measure anxiety- and depressive-like behaviors.
The mutant mice showed fewer anxious behaviors and also demonstrated resistance to the effects of chronic mild, unpredictable stress. Furthermore, even without antidepressants, removing Nf1 from NPCs in adult mice decreased symptoms of depression and anxiety.
“This study demonstrates that inducing neurogenesis is sufficient to produce antidepressant behavioral actions, and provides novel targets for therapeutic interventions,” said Ronald Duman, PhD, a neurogenesis expert from Yale University.
From PsychCentral.com
Tuesday 6 March 2012
Scientists discover that specific antibodies halt Alzheimer's disease in mice
Antibodies that block the process of synapse disintegration in
Alzheimer's disease have been identified, raising hopes for a treatment
to combat early cognitive decline in the disease.
Alzheimer's disease is characterized by abnormal deposits in the
brain of the protein Amyloid-ß, which induces the loss of connections
between neurons, called synapses.
Now, scientists at UCL have discovered that specific antibodies
that block the function of a related protein, called Dkk1, are able to
completely suppress the toxic effect of Amyloid-ß on synapses. The
findings are published today in the Journal of Neuroscience.
Dr Patricia Salinas, from the UCL Department of Cell &
Developmental Biology, who led the study, said: "These novel findings
raise the possibility that targeting this secreted Dkk1 protein could
offer an effective treatment to protect synapses against the toxic
effect of Amyloid-ß.
"Importantly, these results raise the hope for a treatment and perhaps the prevention of cognitive decline early in Alzheimer's disease."
Dkk1 is elevated in the brain biopsies of people with Alzheimer's
disease but the significance of these findings was previously unknown.
Scientists at UCL have found that Amyloid-ß causes the production of
Dkk1, which in turn induces the dismantling of synapses (the connections
between neurons) in the hippocampus, an area of the brain implicated in
learning and memory.
In this paper, scientists conducted experiments to look at the
progression of synapse disintegration of the hippocampus after exposure
to Amyloid-ß, using brain slices from mice. They were able to monitor
how many synapses survived in the presence of a specific antibody which
targets Dkk1, compared to how many synapses were viable without the
antibody.
The results show that the neurons that were exposed to the antibody remained healthy, with no synaptic disintegration.
Dr Salinas said: "Despite significant advances in understanding the
molecular mechanisms involved in Alzheimer's disease, no effective
treatment is currently available to stop the progression of this
devastating disease."
She added: "This research identifies Dkk1 as a potential therapeutic target for the treatment of Alzheimer's disease."
Alzheimer's represents 60% of all cases of dementia. Alzheimer's
Research UK estimates that in the UK the annual national cost of all the
dementias is around £23 billion, which represents double the costs for
cancer and 3-5 times the costs for heart disease and stroke. New studies
predict an increase in the number of Alzheimer's cases of epidemic
proportions.
The research was funded by Alzheimer's Research UK, the UK's leading
dementia research charity, and the Biotechnology and Biological Sciences
Research Council, UK.
Dr Simon Ridley, Head of Research at Alzheimer's Research UK, said:
"We're delighted to have supported this study, which sheds new light on
the processes that occur as Alzheimer's develops. By understanding what
happens in the brain
during Alzheimer's, we stand a better chance of developing new
treatments that could make a real difference to people with the disease.
"Studies like this are an essential part of that process, but more
work is needed if we are to take these results from the lab bench to the
clinic. Dementia can only be defeated through research, and with the
numbers of people affected by the condition soaring, we urgently need to
invest in research now."
Friday 2 March 2012
"SpeechJammer" Invention Stops A Person Talking Mid-Sentence
Two researchers in Japan have invented a "SpeechJammer" device that can stop a person talking in mid-sentence, by just projecting back to them "their own utterances at a delay of a few hundred milliseconds".
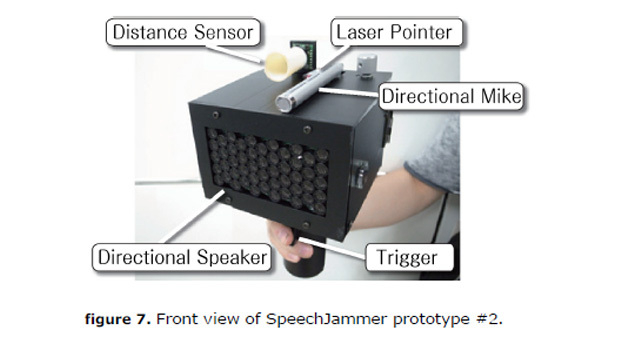 The device does not stop them talking permanently, it is just that they become so confused, they can't finish their sentence and begin to stutter or just shut up.
The device does not stop them talking permanently, it is just that they become so confused, they can't finish their sentence and begin to stutter or just shut up.
The two researchers are Kazutaka Kurihara, a media interaction research scientist at the National Institute of Advanced Industrial Science and Technology, and Koji Tsukada, an assistant professor at Ochanomizu University, and a researcher at JST PRESTO, a program that aims to "cultivate the seeds of precursory science and technology".
They describe their prototype SpeechJammer, and the results of some experiments, in a paper published on 28 February on arVix, an e-print service owned, operated and quality controlled by Cornell University.
The researchers say the device causes no physical discomfort to the interrupted speaker, and the effect stops as soon as they stop speaking.
The prototype SpeechJammer looks like a black cube about the size of a shoebox mounted on a shaft which acts as a handle. The box contains a direction-sensitive speaker, and on top of it is a direction-sensitive microphone.
On Kazutaka Kurihara's personal website there is a short video demonstrating the use of the device in two scenarios.
The first scenario shows a small group of people in an office, working at their computers, when one of them receives a call on her cellphone. The conversation begins to irritate the others, and then one of them decides to take action. He points the SpeechJammer at the irritating talker, interrupting her mid-sentence in her cellphone conversation, whereupon she appears confused, and then stops.
In the other scenario, a lecturer is talking and his lecture has run over time. Many of his students are looking quite bored and fed up and one of them takes the SpeechJammer, points it at the lecturer, and he trips over his own words and stutters, interrupting his flow.
The SpeechJammer works on the principle of Delayed Audio Feedback or DAF. There is a theory that when we speak, we use the sound of our own voice uttering the words to help us. But, if that "playback" is artificially delayed, it interrupts the cognitive processing that helps us maintain our flow. In fact, there is a theory that something akin to DAF is what happens to people who stutter, and it is known that artificially induced DAF can help reduce stuttering.
In their paper the researchers describe how they experimented with two speech contexts: one where the speaker was reading news out loud and another that was a "spontaneous monologue".
It appears that speech jamming is more successful, with this prototype, in the news out loud than in the monologue context, and also, it became obvious that it never works when meaningless sound is uttered, like when someone says "Ahhh" over a long period of time.
With reference to research on communication and decision making, Kurihara and Tsukada point out that applying rules and constraints on verbal contributions can change the properties of the discussion, and they also mention how "negative features" of speech can be "barriers toward peaceful communication".
They propose that using the SpeechJammer to place a constraint on communication, by simply making "speech difficult for some people", it might "bring meaningful changes to communication patterns in discussions".
Such a system "points the way to promising future research relating to discussion dynamics," they write.
In their paper, the researchers focus very much on the science: the physics of the device and how it might be improved to deal with various parameters, plus the science of communication, and make no mention of the ethical and legal aspects of developing a machine that makes people stop talking.
Catharine Paddock PhD. (2012, March 2). ""SpeechJammer" Invention Stops A Person Talking Mid-Sentence." Medical News Today.
The device does not stop them talking permanently, it is just that they become so confused, they can't finish their sentence and begin to stutter or just shut up.The two researchers are Kazutaka Kurihara, a media interaction research scientist at the National Institute of Advanced Industrial Science and Technology, and Koji Tsukada, an assistant professor at Ochanomizu University, and a researcher at JST PRESTO, a program that aims to "cultivate the seeds of precursory science and technology".
They describe their prototype SpeechJammer, and the results of some experiments, in a paper published on 28 February on arVix, an e-print service owned, operated and quality controlled by Cornell University.
The researchers say the device causes no physical discomfort to the interrupted speaker, and the effect stops as soon as they stop speaking.
The prototype SpeechJammer looks like a black cube about the size of a shoebox mounted on a shaft which acts as a handle. The box contains a direction-sensitive speaker, and on top of it is a direction-sensitive microphone.
On Kazutaka Kurihara's personal website there is a short video demonstrating the use of the device in two scenarios.
The first scenario shows a small group of people in an office, working at their computers, when one of them receives a call on her cellphone. The conversation begins to irritate the others, and then one of them decides to take action. He points the SpeechJammer at the irritating talker, interrupting her mid-sentence in her cellphone conversation, whereupon she appears confused, and then stops.
In the other scenario, a lecturer is talking and his lecture has run over time. Many of his students are looking quite bored and fed up and one of them takes the SpeechJammer, points it at the lecturer, and he trips over his own words and stutters, interrupting his flow.
The SpeechJammer works on the principle of Delayed Audio Feedback or DAF. There is a theory that when we speak, we use the sound of our own voice uttering the words to help us. But, if that "playback" is artificially delayed, it interrupts the cognitive processing that helps us maintain our flow. In fact, there is a theory that something akin to DAF is what happens to people who stutter, and it is known that artificially induced DAF can help reduce stuttering.
In their paper the researchers describe how they experimented with two speech contexts: one where the speaker was reading news out loud and another that was a "spontaneous monologue".
It appears that speech jamming is more successful, with this prototype, in the news out loud than in the monologue context, and also, it became obvious that it never works when meaningless sound is uttered, like when someone says "Ahhh" over a long period of time.
With reference to research on communication and decision making, Kurihara and Tsukada point out that applying rules and constraints on verbal contributions can change the properties of the discussion, and they also mention how "negative features" of speech can be "barriers toward peaceful communication".
They propose that using the SpeechJammer to place a constraint on communication, by simply making "speech difficult for some people", it might "bring meaningful changes to communication patterns in discussions".
Such a system "points the way to promising future research relating to discussion dynamics," they write.
In their paper, the researchers focus very much on the science: the physics of the device and how it might be improved to deal with various parameters, plus the science of communication, and make no mention of the ethical and legal aspects of developing a machine that makes people stop talking.
Catharine Paddock PhD. (2012, March 2). ""SpeechJammer" Invention Stops A Person Talking Mid-Sentence." Medical News Today.
Lead Interferes With The Synthesis And Function Of Brain-Derived Neurotropic Factor, Derailing The Brain's Center For Learning
Exposure to lead wreaks havoc in the brain, with consequences that include lower IQ and reduced potential for learning. But the precise mechanism by which lead alters nerve cells in the brain has largely remained unknown.
New research led by Tomás R. Guilarte, PhD, Leon Hess Professor and Chair of Environmental Health Sciences at Columbia University Mailman School of Public Health, and post-doctoral research scientist Kirstie H. Stansfield, PhD, used high-powered fluorescent microscopy and other advanced techniques to painstakingly chart the varied ways lead inflicts its damage. They focused on signaling pathways involved in the production of brain-derived neurotropic factor, or BDNF, a chemical critical to the creation of new synapses in the hippocampus, the brain's center for memory and learning.
The study appears online in the journal Toxicological Sciences.
Once BDNF is produced in the nucleus, explains Dr. Stansfield, it is transported as cargo in a railroad-car-like vesicle along a track called a microtubule toward sites of release in the axon and dendritic spines. Vesicle navigation is controlled in part through activation (phosphorylation) of the huntingtin protein, which as its name suggests, was first identified through research into Huntington's disease. By looking at huntingtin expression, the researchers found that lead exposure, even in small amounts, is likely to impede or reverse the train by altering phosphorylation at a specific amino acid.
The BDNF vesicle transport slowdown is just one of a variety of ways that lead impedes BDNF's function. The researchers also explored how lead curbs production of BDNF in the cell nucleus. One factor, they say, may be a protein called methyl CpG binding protein 2, or MeCP2, which has been linked with RETT syndrome and autism spectrum disorders and acts to "silence" BDNF gene transcription.
The paper provides the first comprehensive working model of the ways by which lead exposure impairs synapse development and function. "Lead attacks the most fundamental aspect of the brain - the synapse. But by better understanding the numerous and complex ways this happens we will be better able to develop therapies that ameliorate the damage," says Dr. Guilarte.
New research led by Tomás R. Guilarte, PhD, Leon Hess Professor and Chair of Environmental Health Sciences at Columbia University Mailman School of Public Health, and post-doctoral research scientist Kirstie H. Stansfield, PhD, used high-powered fluorescent microscopy and other advanced techniques to painstakingly chart the varied ways lead inflicts its damage. They focused on signaling pathways involved in the production of brain-derived neurotropic factor, or BDNF, a chemical critical to the creation of new synapses in the hippocampus, the brain's center for memory and learning.
The study appears online in the journal Toxicological Sciences.
Once BDNF is produced in the nucleus, explains Dr. Stansfield, it is transported as cargo in a railroad-car-like vesicle along a track called a microtubule toward sites of release in the axon and dendritic spines. Vesicle navigation is controlled in part through activation (phosphorylation) of the huntingtin protein, which as its name suggests, was first identified through research into Huntington's disease. By looking at huntingtin expression, the researchers found that lead exposure, even in small amounts, is likely to impede or reverse the train by altering phosphorylation at a specific amino acid.
The BDNF vesicle transport slowdown is just one of a variety of ways that lead impedes BDNF's function. The researchers also explored how lead curbs production of BDNF in the cell nucleus. One factor, they say, may be a protein called methyl CpG binding protein 2, or MeCP2, which has been linked with RETT syndrome and autism spectrum disorders and acts to "silence" BDNF gene transcription.
The paper provides the first comprehensive working model of the ways by which lead exposure impairs synapse development and function. "Lead attacks the most fundamental aspect of the brain - the synapse. But by better understanding the numerous and complex ways this happens we will be better able to develop therapies that ameliorate the damage," says Dr. Guilarte.
Columbia University's Mailman School of Public Hea. (2012, March 2). "Lead Interferes With The Synthesis And Function Of Brain-Derived Neurotropic Factor, Derailing The Brain's Center For Learning." Medical News Today.
Restricting Enzyme Reverses Alzheimer's Symptoms In Mice
A study conducted by Li-Huei Tsai, a researcher at MIT, has found that an enzyme (HDAC2) overproduced in the brains of individuals with Alzheimer's, blocks genes needed to develop new memories. With this finding, the team were able to restrict this enzyme in mice and reverse symptoms of Alzheimer's. Results from the study are published in the February 29 online edition of Nature.
Alzheimer's currently affects 5.4 million people in the United States. Findings from the study indicate that medications targeting HDAC2 could be a new techniques to treating Alzheimer's. Globally, the incidence of people with Alzheimer's is expected to increase two fold every two decades. Recently, President Barack Obama set a goal date of 2025 to find an effective treatment.
According to Tsai, this goal could be achieved with the help of HDAC2 inhibitors, although it would probably take a minimum of at least a decade in order to develop and test such medications. Lead author of the report is Johannes Gräff, a postdoc at the Picower Institute.
Tsai, director of the Picower Institute for Learning and Memory at MIT, explained:
Histone deacetylases (HDACs) are a class of 11 enzymes that control gene regulation by altering histones, which consist of highly alkaline proteins and are the chief protein components of chromatin. Histones act as spools around which DNA winds and play a role in gene regulation. HDACs transform a histone via a method called deacetylation. During this process, chromatin is packaged more tightly, making gens in that area less likely to be expressed.
This effect can be reversed using HDAC inhibitors. These inhibitors open up the DNA and allow it to be transcribed.
In prior investigations, Tsai has demonstrated the HDAC2 is an important regulator of memory and learning. In this study, the team found that restricting HDAC2 can reverse symptoms of Alzheimer's in rodents.
The team discovered that HDAC2, yet no other HDACs, is overproduced in the hippocampus of mice with Alzheimer's symptoms. The hippocampus is a region in the brain where new memories are created.
HDAC2 was most frequently found attached to genes involved in synaptic plasticity. Synaptic plasticity is the brains ability change the connection strength between two neurons, in response to new information, which is vital to making memories.
In addition, the researchers found that those genes had significantly lower levels of acetylation and expression in the affected mice.
Tsai explains:
Using short hairpin RNA, a molecule which can develope to attach to a carrier RNA, the team blocked HDAC2 in the hippocampi of mice with Alzheimer's symptoms. RNA is a molecule that delivers genetic instructions from DNA to the rest of the cell.
The researchers found that reduced HDAC2 activity restarted histone acetylation, allowing genes needed for synaptic plasticity and other memory and learning processes to be expressed. They discovered that synaptic density increased considerably in treated mice, and that the rodents regained normal cognitive function.
Tsai, said:
In addition, the team evaluated postmortem brains of Alzheimer's patients and discovered increased levels of HDAC2 in the hippocampus and entorhinal cortex, which play vital roles in memory storage.
According to Tsai, results from the study may explain why medications that remove beta-amyloid proteins from the brains of Alzheimer's patients have only provided modest, if any, improvements in human trials.
In the brains of Alzheimer's patients, beta-amyloid proteins are known to clump. This clumping interferes with a type of cell receptor required for synaptic plasticity. Results from the study demonstrate that beta-amyloid also activates the generation of HDAC2, possibly initiating the restriction of memory and learning genes.
Tsai explains:
"We think that once this epigenetic blockage of gene expression is in place, clearing beta amyloid may not be sufficient to restore the active configuration of the chromatin."
According to Tsai, HDAC2 inhibitors are appealing, as they could possibly reverse Alzheimer's symptoms even after the blockage is well-established, although significantly more medication development needs to be conducted before using such drugs in human trials.
Tsai says:
"It's really hard to predict. Clinical trials would probably be five years down the line. And if everything goes well, to become an approved drug would probably take at least 10 years."
Although some researchers have tested some general HDAC inhibitors, not specific to HDAC2, in human trials as cancer medications, a more selective approach is required to treat Alzheimer's. Tsai explains:
"You want something as selective as possible, and as safe as possible."
Alzheimer's currently affects 5.4 million people in the United States. Findings from the study indicate that medications targeting HDAC2 could be a new techniques to treating Alzheimer's. Globally, the incidence of people with Alzheimer's is expected to increase two fold every two decades. Recently, President Barack Obama set a goal date of 2025 to find an effective treatment.
According to Tsai, this goal could be achieved with the help of HDAC2 inhibitors, although it would probably take a minimum of at least a decade in order to develop and test such medications. Lead author of the report is Johannes Gräff, a postdoc at the Picower Institute.
Tsai, director of the Picower Institute for Learning and Memory at MIT, explained:
"I would really strongly advocate for an active program to develop agents that can contain HDAC2 activity. The disease is so devastating and affects so many people, so I would encourage more people to think about this."
Histone deacetylases (HDACs) are a class of 11 enzymes that control gene regulation by altering histones, which consist of highly alkaline proteins and are the chief protein components of chromatin. Histones act as spools around which DNA winds and play a role in gene regulation. HDACs transform a histone via a method called deacetylation. During this process, chromatin is packaged more tightly, making gens in that area less likely to be expressed.
This effect can be reversed using HDAC inhibitors. These inhibitors open up the DNA and allow it to be transcribed.
In prior investigations, Tsai has demonstrated the HDAC2 is an important regulator of memory and learning. In this study, the team found that restricting HDAC2 can reverse symptoms of Alzheimer's in rodents.
The team discovered that HDAC2, yet no other HDACs, is overproduced in the hippocampus of mice with Alzheimer's symptoms. The hippocampus is a region in the brain where new memories are created.
HDAC2 was most frequently found attached to genes involved in synaptic plasticity. Synaptic plasticity is the brains ability change the connection strength between two neurons, in response to new information, which is vital to making memories.
In addition, the researchers found that those genes had significantly lower levels of acetylation and expression in the affected mice.
Tsai explains:
"It's not just one or two genes, it's a group of genes that work in concert to control different phases of memory formation. Which such a blockade, the brain really loses the ability to quickly respond to stimulation. You can imagine that this creates a huge problem in terms of learning and memory functions, and perhaps other cognitive functions."
Using short hairpin RNA, a molecule which can develope to attach to a carrier RNA, the team blocked HDAC2 in the hippocampi of mice with Alzheimer's symptoms. RNA is a molecule that delivers genetic instructions from DNA to the rest of the cell.
The researchers found that reduced HDAC2 activity restarted histone acetylation, allowing genes needed for synaptic plasticity and other memory and learning processes to be expressed. They discovered that synaptic density increased considerably in treated mice, and that the rodents regained normal cognitive function.
Tsai, said:
"This result really advocates for the notion that if there is any agent that can selectively down-regulate HDAC2, it's going to be very beneficial."
In addition, the team evaluated postmortem brains of Alzheimer's patients and discovered increased levels of HDAC2 in the hippocampus and entorhinal cortex, which play vital roles in memory storage.
According to Tsai, results from the study may explain why medications that remove beta-amyloid proteins from the brains of Alzheimer's patients have only provided modest, if any, improvements in human trials.
In the brains of Alzheimer's patients, beta-amyloid proteins are known to clump. This clumping interferes with a type of cell receptor required for synaptic plasticity. Results from the study demonstrate that beta-amyloid also activates the generation of HDAC2, possibly initiating the restriction of memory and learning genes.
Tsai explains:
"We think that once this epigenetic blockage of gene expression is in place, clearing beta amyloid may not be sufficient to restore the active configuration of the chromatin."
According to Tsai, HDAC2 inhibitors are appealing, as they could possibly reverse Alzheimer's symptoms even after the blockage is well-established, although significantly more medication development needs to be conducted before using such drugs in human trials.
Tsai says:
"It's really hard to predict. Clinical trials would probably be five years down the line. And if everything goes well, to become an approved drug would probably take at least 10 years."
Although some researchers have tested some general HDAC inhibitors, not specific to HDAC2, in human trials as cancer medications, a more selective approach is required to treat Alzheimer's. Tsai explains:
"You want something as selective as possible, and as safe as possible."
Grace Rattue. (2012, February 29). "Restricting Enzyme Reverses Alzheimer's Symptoms In Mice." Medical News Today.
Thursday 1 March 2012
Activating The Visual Cortex Improves Our Sense Of Smell
A new study reveals for the first time that activating the brain's visual cortex with a small amount of electrical stimulation actually improves our sense of smell.
The finding published in the Journal of Neuroscience by researchers at the Montreal Neurological Institute and Hospital - The Neuro, McGill University and the Monell Chemical Senses Center, Philadelphia, revises our understanding of the complex biology of the senses in the brain.
"It's known that there are separate specialized brain areas for the different senses such as vision, smell, touch and so forth but, when you experience the world around you, you get a coherent picture based on information from all the senses. We wanted to find out how this works in the brain," says Dr. Christopher Pack, lead investigator at The Neuro. "In particular we wanted to test the idea that activation of brain regions primarily dedicated to one sense might influence processing in other senses. What we found was that electrically stimulating the visual cortex improves performance on a task that requires participants to identify the odd odor out of a group of three." This result is interesting because it shows, for the first time, that on a basic level the brain structures involved in different senses are really quite interconnected in everyone - more so than previously understood.
"This 'cross-wiring' of senses has been described in people with synesthesia, a condition in which stimulation of one sense leads to automatic, involuntary experiences in a second sense, causing people to see the colour of numbers, or smell words, or hear odours for example, says Dr. Johan Lundstrom at Monell Chemical Senses Center. "Now this study shows that cross-wiring of the senses exists in all of us, so we could all be considered synesthetic to a degree."
To examine the possibility that activating the visual cortex influences the sense of smell, people were tested on smell tasks before and after application of TMS, a non-invasive method of stimulating targeted brain areas. TMS, or transcranial magnetic stimulation, was directed towards the visual cortex using a protocol that had been previously shown by researchers at The Neuro to improve visual perception. TMS is already widely used in the treatment of certain disease symptoms, and because TMS alters brain activity in a targeted area, it provides a powerful test of the hypothesis that visual cortex activation changes olfactory perception.
The results demonstrate that visual cortex activity is incorporated into the processing of smells, proving for the first time a cross-wiring of the visual and olfactory systems in the brain. Interestingly, the team did not find evidence for similar cross-wiring between olfactory and auditory systems. This suggests that vision may play a special role in binding together information from the different senses, a possibility that the researchers are currently exploring. In addition to Drs. Pack and Lundstrom, the research was carried out by Jahan Jadauji, a Master's student, and Jelena Djordjevic, a clinical neuropsychologist and neuroscientist, both at The Neuro. This collaboration between researchers and clinicians was made possible by The Neuro's integrated research institute and hospital.
From Medical News Today
"It's known that there are separate specialized brain areas for the different senses such as vision, smell, touch and so forth but, when you experience the world around you, you get a coherent picture based on information from all the senses. We wanted to find out how this works in the brain," says Dr. Christopher Pack, lead investigator at The Neuro. "In particular we wanted to test the idea that activation of brain regions primarily dedicated to one sense might influence processing in other senses. What we found was that electrically stimulating the visual cortex improves performance on a task that requires participants to identify the odd odor out of a group of three." This result is interesting because it shows, for the first time, that on a basic level the brain structures involved in different senses are really quite interconnected in everyone - more so than previously understood.
"This 'cross-wiring' of senses has been described in people with synesthesia, a condition in which stimulation of one sense leads to automatic, involuntary experiences in a second sense, causing people to see the colour of numbers, or smell words, or hear odours for example, says Dr. Johan Lundstrom at Monell Chemical Senses Center. "Now this study shows that cross-wiring of the senses exists in all of us, so we could all be considered synesthetic to a degree."
To examine the possibility that activating the visual cortex influences the sense of smell, people were tested on smell tasks before and after application of TMS, a non-invasive method of stimulating targeted brain areas. TMS, or transcranial magnetic stimulation, was directed towards the visual cortex using a protocol that had been previously shown by researchers at The Neuro to improve visual perception. TMS is already widely used in the treatment of certain disease symptoms, and because TMS alters brain activity in a targeted area, it provides a powerful test of the hypothesis that visual cortex activation changes olfactory perception.
The results demonstrate that visual cortex activity is incorporated into the processing of smells, proving for the first time a cross-wiring of the visual and olfactory systems in the brain. Interestingly, the team did not find evidence for similar cross-wiring between olfactory and auditory systems. This suggests that vision may play a special role in binding together information from the different senses, a possibility that the researchers are currently exploring. In addition to Drs. Pack and Lundstrom, the research was carried out by Jahan Jadauji, a Master's student, and Jelena Djordjevic, a clinical neuropsychologist and neuroscientist, both at The Neuro. This collaboration between researchers and clinicians was made possible by The Neuro's integrated research institute and hospital.
From Medical News Today
New drug offers bigger window to treat stroke
A DRUG which minimises brain damage when given three hours after stroke has proved successful in monkeys and humans.
A lack of oxygen in the brain during a stroke can cause fatal brain damage. There is only one approved treatment - tissue plasminogen activator - but it is most effective when administered within 90 minutes after the onset of stroke. Immediate treatment isn't always available, however, so drugs that can be given at a later time have been sought.
In a series of experiments, Michael Tymianski and colleagues at Toronto Western Hospital in Ontario, Canada, replicated the effects of stroke in macaques before intravenously administering a PSD-95 inhibitor, or a placebo. PSD-95 inhibitors interfere with the process that triggers cell death when the brain is deprived of oxygen.
To test its effectiveness the team used MRI to measure the volume of damaged brain for 30 days following the treatment, and conducted behavioural tests at various intervals within this time.
Monkeys treated with the PSD-95 inhibitor one hour after stroke had 55 per cent less damaged tissue in the brain after 24 hours and 70 per cent less after 30 days, compared with those that took a placebo. These animals also did better in behavioural tests. Importantly, the drug was also effective three hours after stroke (Nature, DOI: 10.1038/nature10841).
From NewScientist
Subscribe to:
Posts (Atom)
Popular Posts
-
 A recent BBC drama portrayed a young, psychopathic Russian assassin named ‘Villanelle’ – supposedly derived from the word “villainess”. Bas...
A recent BBC drama portrayed a young, psychopathic Russian assassin named ‘Villanelle’ – supposedly derived from the word “villainess”. Bas... -
 In 1935, Alcoholics Anonymous was founded, with the goal of understanding and treating the “intrapsychic” and social forces causing addictio...
In 1935, Alcoholics Anonymous was founded, with the goal of understanding and treating the “intrapsychic” and social forces causing addictio... -
There seems to be a phenomenon prevalent across much of Western society. People don't like to accept responsibility for their own action...
-
When you experience a new event, your brain encodes a memory of it by altering the connections between neurons. This requires turning on...
-
Accepted model for brain signaling flawed @ ScienceDaily A new study out January 10 in the journal Science turns two decades of under...
-
 A study published yesterday in Cell [1] has purportedly found that memories formed as infants may be able to be retrieved as adults using o...
A study published yesterday in Cell [1] has purportedly found that memories formed as infants may be able to be retrieved as adults using o...
